La conmemoración de este día llega en un año muy particular, marcado por la reciente aprobación de la Ley de FQ, y con la certeza de que este gran logro será un importante avance para todos los pacientes. Para hacer una puesta al día sobre esta enfermedad las Doctoras Laura Osken (Procoordinadora), Silvina Smith (Vocal) y Patricia Andreozzi (Coordinadora) de la Sección de Fibrosis Quística de la AAMR, nos invitan a leer un artículo científico elaborado especialmente para esta gaceta.
INTRODUCCIÓN
La Fibrosis Quística (FQ) es una enfermedad hereditaria autosómica recesiva grave más frecuente en la población caucásica causada por mutaciones en el gen localizado en el brazo largo del cromosoma 7. La primera descripción de la FQ como entidad fue realizada por Andersen en 1938. Recién en la década de 1980 se descubrió que el defecto fundamental se debe al intercambio alterado del ion cloro de las glándulas de secreción exocrinas. En 1989 se logró el aislamiento y caracterización del gen responsable que codifica la proteína reguladora de la conductancia transmembrana (CFTR) que actúa como canal principal del cloro de la membrana e influye en otros iones (calcio, sodio, etc.). Este gen provoca una alteración en la producción o funcionamiento normal de la proteína CFTR que se encuentra en la membrana apical del epitelio secretor de las glándulas mucosas de vías aéreas, digestivas, reproductor y sudor.
En Argentina aproximadamente la incidencia de la enfermedad es de 1/6700 recién nacidos y de estos, alrededor del 60.4% tienen diagnóstico antes del primer año de vida, en donde juega un rol importante la pesquisa neonatal (Ley nacional 26279). La instauración de un tratamiento adecuado en forma precoz, los conocimientos de la fisiopatología y la atención por un equipo especializado permiten disminuir la progresión de la enfermedad y aumentar la sobrevida, calculada en alrededor de los 40 años en los países desarrollados. La prevalencia de portadores sanos de la mutación es aproximadamente 1/40.
Los pacientes con FQ deben tener dos mutaciones causantes de la enfermedad en trans (en distintos cromosomas) en el gen CFTR localizado en el cromosoma 7. En este momento, hay alrededor de más de 2000 mutaciones conocidas que afectan al CFTR registradas en la Cystic Fibrosis Mutation Database (CFMD) muchas de las cuales dan lugar a un fenotipo patológico. Las mutaciones se clasifican de acuerdo con el grado de alteración y síntesis del CFTR. Alrededor del 75% de los alelos FQ contienen la mutación F508del. Esta base de datos ha servido de referencia para la clasificación de las mutaciones.
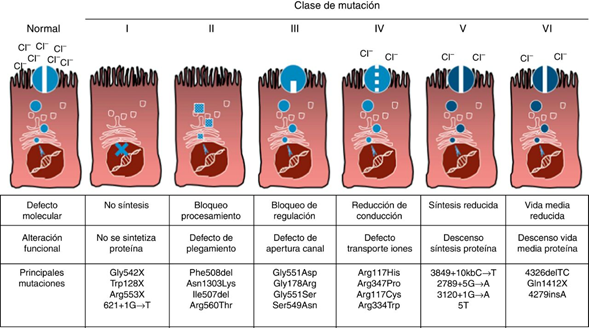
Clase 1: Proteína CFTR no funcional (síntesis truncada)
Clase 2: Proteína CFTR poco funcional (defecto en el procesamiento y transporte intracelular)
Clase 3: Defecto en la regulación del transporte de cloruros.
Clase 4: Defecto de la conductancia del canal de cloruros con alguna funcionalidad del CFTR
Clase 5: Cantidad disminuida de proteína CFTR funcional
Clase 6: Disminución de la estabilidad de la proteína CFTR
Las clases 1, 2 y 3 están asociadas a escasa o nula función del CFTR y por consiguiente con cuadros más severos de la enfermedad, y como expresión clínica incluyen aquellos casos con insuficiencia pancreática exocrina. Las clases 4 y 5 incluyen variantes con función residual del CFTR. Algunas mutaciones como la F508del (clase2) cumple criterios de más de una clase funcional, siendo también de clase 3 y 6.
El concepto actual de la patogénesis de la enfermedad pulmonar en FQ consiste en un transporte anormal de Cloro y de otros iones como el Na, Ca y agua a través de la membrana apical, que lleva a la deshidratación de la capa de líquido de su superficie y alteración del aclaramiento mucociliar. Las secreciones se vuelven viscosas y obstruyen las vías aéreas y alteran la eliminación de bacterias permitiendo que se establezca la infección bacteriana, que conduce a inflamación, preferentemente neutrofílica y el desarrollo de bronquiectasias. El compromiso respiratorio es la principal causa de morbimortalidad.
Las manifestaciones más frecuentes de la enfermedad son la insuficiencia pancreática exocrina en alrededor del 85-90% de los casos, la enfermedad pulmonar obstructiva crónica grave característica, que se desarrolla con el tiempo en casi todos los casos, la azoospermia obstructiva por anomalías anatómicas en el tracto urogenital en la casi totalidad de los pacientes masculinos y altas concentraciones de cloro y sodio en el sudor en más del 98% de los casos.
La presentación clínica es muy variable, desde la clásica grave con síntomas malabsortivos y respiratorios crónicos desde poco después del nacimiento, a fenotipos leves como infertilidad, síntomas debidos a las perdidas excesivas de sal por el sudor, o poliposis nasal.
Los criterios clásicos de la enfermedad son: 1 criterio clínico y una prueba que determine la disfunción del CFTR, siendo las más importante la Prueba o Test del Sudor (test cuantitativo de iontoforesis por pilocarpina) superior a 60 mmol/L.
DIAGNOSTICO:
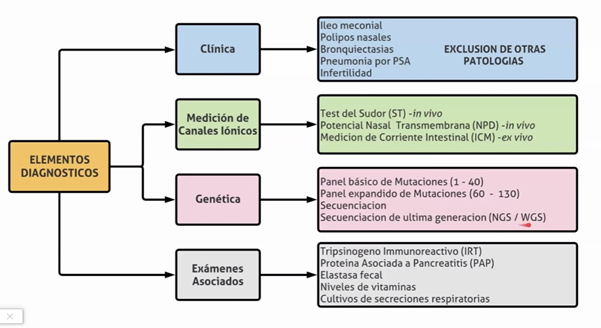
La enfermedad puede sospecharse a través de una pesquisa neonatal positiva, por antecedente de un hermano afectado o por manifestaciones clínicas y se confirma detectando la disfunción del canal CFTR mediante la prueba de sudor positiva y/o el hallazgo de dos mutaciones del gen CFTR causantes de FQ.
Pesquisa neonatal (PN): es obligatoria en nuestro país como lo indica la Ley nacional 26.279 (año 2007), con la determinación de la Tripsina Inmuno Reactiva (TIR) la que esta elevada en los afectados, debe realizarse antes de las 48hs. y repetirse alrededor de la segunda y tercera semana de vida, porque luego los valores descienden, y podemos tener falsos negativos. En los prematuros deberá repetirse a las 37 semanas. En caso de ser igual o mayor de 70 ng/dl se repite la determinación (método TIR/TIR), y si esta segunda determinación es anormal se debe solicitar la prueba del sudor para confirmar el diagnóstico.
Prueba del sudor. El método cuantitativo desarrollado por Gibson y Cooke para la determinación de cloruros: sigue siendo el método diagnóstico más sensible.
Igual o mayor a 60 mmol/ (confirman la enfermedad).
Valores intermedios entre 30 a 59 mmol/L son dudosos debería repetirse la prueba y con valores por debajo de 30 mmol/L, la FQ es improbable (excepto algunas mutaciones).
Ante un niño con PN positiva y valores de cloro en sudor intermedios (en al menos dos oportunidades) y en los que el estudio molecular habitual ha detectado menos de 2 mutaciones causantes de FQ (1 o ninguna), se debe realizar un estudio ampliado de mutaciones (secuenciación del gen CFTR) u otros métodos de evaluación de la función del CFTR como la Diferencia de Potencial Nasal (DPN). Estos estudios están limitados al médico especialista en FQ, por lo tanto, estos pacientes deben ser derivados a centros especializados en Fibrosis Quística.
Son muy pocos los centros en donde se pueden realizar este tipo de estudio (DPN) para demostrar la anormal función del CFTR para los que se requiere amplia experiencia y hay consenso en que este método debe reservarse para los pacientes con diagnostico dudoso. En caso de no poder confirmar el diagnóstico el paciente debe ser asistido igualmente en un centro de FQ.
TRATAMIENTO:
La esperanza de vida en la FQ ha mejorado sustancialmente en las últimas décadas, tanto sea por las nuevas terapias, la atención en centros especializados y la aplicación de la medicina personalizada. Sin embargo, la mayoría fallecen por insuficiencia respiratoria, por lo tanto, la disminución de la progresión de la enfermedad pulmonar es un objetivo principal en el tratamiento.
La infección crónica está marcada por exacerbaciones agudas, después de los cual la función pulmonar puede no volver a los niveles basales, estas marcan el pronóstico de la enfermedad, ya que a mayor número de estas se produce una disminución en la expectativa de vida.
Los tres pilares fundamentales en el tratamiento son: mantener un estado nutricional adecuado, que favorece una mejor función pulmonar, la higiene bronquial con técnicas de kinesiología y dispositivos más en las infecciones respiratorias la antibióticoterapia. El tratamiento del resto de las manifestaciones clínicas y las nuevas terapias también son fundamentales en el tratamiento de la FQ.
Mucolíticos: Dornasa alfa produce la degradación del moco generado principalmente por la degradación de neutrófilos.
Terapia Osmótica: Solución salina hipertónica y Manitol producen la hidratación del moco por arrastre del agua a la superficie celular.
Inflamación: Azitromicina probable mecanismo en la reducción de la formación de Biofilms en las infecciones por Pseudomonas aeruginosa.
Tratamiento de las infecciones: Antibióticos, la elección de antibióticos es clave para erradicar nuevas bacterias, evitar exacerbaciones y para terapias de mantenimiento. La premisa de ATB inhalados es apuntar al sitio de la infección y disminuir la densidad bacteriana del esputo, mientras que los efectos secundarios sistémicos son mínimos.
Moduladores: Son de reciente aparición, diferentes tipos según mecanismo de acción se pueden usar como terapia única, doble terapia o triple terapia según las variantes genéticas halladas en el paciente y la edad del mismo. Deben ser referenciadas por especialistas en FQ.
Potenciadores: activan el CFTR permitiendo su actividad de regular el influjo de Cl a través del canal.
Correctores: reparan el procesamiento defectuoso facilitando la maduración apropiada y suministro de proteína a la membrana.
Amplificadores: aumentan selectivamente la disponibilidad del CFTR inmaduro.
Nutrición: es uno de los pilares fundamentales en el tratamiento de los pacientes con FQ, una buena nutrición también aumenta la calidad y expectativa de vida del paciente, en esta incluimos: Enzimas pancreáticas, Vitaminas, Minerales, Zinc y Soporte nutricional vía oral o enteral.
Kinesiología: es esencial tanto en la terapia de mantenimiento como en las exacerbaciones.
Ejercicio: Mejora el componente cardiorrespiratorio, el componente muscular y la composición corporal
Trasplante pulmonar: en situación avanzada de la enfermedad, se recomienda la consulta precoz en pacientes que reúnan criterios a los centros de referencia de trasplante pulmonar del país.
COMPLICACIONES:
Los pacientes con FQ pueden desarrollar una variedad de complicaciones poco frecuentes, pero que el equipo debe estar bien preparado para su correcto diagnóstico y tratamiento.
Las mismas pueden ser:
Digestivas:
-Desnutrición, Diabetes, Enfermedad Hepática, Deshidratación, Reflujo, Cáncer colorrectal
Respiratorias:
-Rinosinusitis, Pólipos nasales, ABPA, Bronquitis Aspergilar, Infección por Micobacterias Atípicas, Hemoptisis, Neumotórax, Atelectasias.
CONCLUSIONES:
El aumento de la supervivencia en FQ es el resultado de varios factores: un diagnóstico precoz, la atención multidisciplinaria y la medicina cada vez más personalizada.
Pero también se ha asociado con la aparición de complicaciones, como el aumento de frecuencia en la resistencia a infecciones y toxicidad asociada a ciertos fármacos.
Una nueva era con los Moduladores y nuevos tratamientos en fase de investigación se imponen, hay un largo camino por recorrer, hay varios factores en los que debemos focalizar como la clase de mutaciones, perfil genético, interacciones farmacológicas, efectos adversos y costo de la medicación para lo cual debemos estar preparados para trabajar con esta visión de futuro, logrando disminuir la progresión de la enfermedad y en consecuencia evitar llegar al trasplante.
Bibliografía
1-Nelson L Turcios. Cystic Fibrosis Lung Disease: An Overview. Respiratory Care February 2020, 65 (2) 233-251. 2-David A. Stoltz, M.D., Ph.D., David K. Meyerholz, D.V.M., Ph.D., and Michael J. Welsh, M.D-Origins of Cystic Fibrosis Lung Disease. N Engl J Med 2015; 372:351-62.