
Autores: Beatriz Gil, Marcela Usandivaras, Juan I. Enghelmayer, María Otaola, Brenda Varela, Adrián Gaser, Gabriela Tabaj, Norma Naval, Silvia Quadrelli.
- Introducción
Algunos pacientes con Enfermedades Pulmonares Intersticiales Difusas (EPID), incluyendo la Fibrosis Pulmonar Idiopática (FPI), pueden evolucionar con un fenotipo fibrosante progresivo caracterizado por un incremento de la extensión de la fibrosis en la Tomografía Computarizada de Alta Resolución (TCAR), declinación de la función pulmonar, empeoramiento tanto de los síntomas respiratorios como de la calidad de vida y mortalidad temprana a pesar de la medicación habitual [1,2]. Se estima que entre un 18-32% de los pacientes con EPID fibrosantes no FPI pueden evolucionar hacia un fenotipo progresivo (EPID-FP).
No existe un consenso en cuanto a la definición de cómo debe ser determinada la progresión de la enfermedad. Ensayos clínicos y estudios observacionales, a pesar de la variabilidad que presenta, en general definen progresión en términos de una declinación de la Capacidad Vital Forzada (CVF), medida como cambios en mililitros (ml) desde el basal, o como porcentaje del valor predicho o como un cambio categórico ≥ 10% del predicho. Esta declinación de la CVF es predictor de mortalidad en pacientes con FPI [3].
Para otras enfermedades fibrosantes (como la enfermedad intersticial asociada a esclerodermia) son predictores de progresión de enfermedad los factores demográficos como el sexo masculino, el origen africano, el fenotipo de la enfermedad como la forma de presentación difusa de la enfermedad o la presencia del anticuerpo anti-sclero70 positivo. Además de la esclerodermia, para otras entidades fibrosantes son predictivos de progresión, la extensión de la fibrosis en las imágenes, el patrón de Neumonía Intersticial Usual (NIU) o la presencia de panalización en la TCAR y una reserva respiratoria disminuida tanto de la CVF como de la Difusión de Monóxido de Carbono (DLCO). Otros factores asociados son la progresión a pesar de la terapia de primera línea de elección o la presencia de una exacerbación aguda u hospitalización por compromiso respiratorio.
La FPI es por definición, una enfermedad fibrosante crónica. Sin embargo, existen otras EPID fibrosantes crónicas que pueden tener riesgo de producir un fenotipo fibrosante [4]. Estas incluyen la Neumonía Intersticial No Específica (NINE), ciertas EPID autoinmunes, sarcoidosis crónica, neumonitis por hipersensibilidad crónica o ciertas enfermedades por exposición como la asbestosis y la silicosis. (Fig.1)
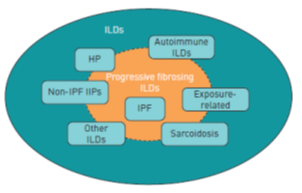
Figura 1. Tipos de EPID que pueden evolucionar al fenotipo fibrosante crónico [4]
El dilema de los «desglosadores vs agrupadores»
Sabemos entonces que la fibrosis pulmonar constituye la etapa final de una amplia gama de enfermedades pulmonares intersticiales heterogéneas y se asocia con deterioro en la calidad de vida, disminución del estado funcional y mortalidad temprana [4,5, 6]. Se caracteriza por destrucción del parénquima pulmonar, depósitos de matriz extracelular y cambios dramáticos en el fenotipo de los fibroblastos y las células alveolo-epiteliales [6,7]. Pertenece al grupo de enfermedades fibroproliferativas que, en conjunto, representan muchas muertes en los países desarrollados [7].
Como veremos más adelante, desde la introducción de los dos fármacos antifibróticos (pirfenidona y nintedanib) en el tratamiento de la FPI, ha habido mucho interés en saber si estos son efectivos en otras enfermedades pulmonares intersticiales fibróticas progresivas (EPID-FP), incluida la neumonitis por hipersensibilidad crónica fibrosante (NHC), EPID asociadas a la enfermedad del tejido conectivo (EPID-ETC) y formas no clasificables de fibrosis pulmonar (EPID nC) [8].
Clasificar la FPI separada de otras, individualizando cada vez más las EPID, nos ha enseñado que desafortunadamente los trastornos progresivos del fenotipo fibrótico no FPI se presentan con un desenlace muy similar a la FPI, especialmente cuando el patrón NIU está presente en la biopsia o en las imágenes por TCAR.
A raíz de estos temas, algunos expertos en EPID/FPI que no solo estudian y difunden estos conocimientos, sino que generan saberes nuevos incesantemente se encuentran, podríamos decir, mínimamente «confrontados».
Aparecen por un lado, los “Desglosadores” y su Medicina de precisión. La medicina de precisión se basa en gran parte, en el descubrimiento de biomarcadores confiables y reproducibles. Son muchos los que a lo largo de estos años han desarrollado esta idea [9, 10,11]. En este contexto específico, los biomarcadores podrían definirse como indicadores medidos objetivamente de mecanismos subyacentes de la enfermedad dados por la combinación de enfoques “ómicos” (ej. genómicos, transcriptómicos, metabolómicos, proteinómicos y microbiomicos) proteínas específicas y/o sus modificaciones en plasma circulante y en el análisis de muestras de tejido pulmonar [12, 13,14]. Hasta el día de hoy son pocos los estudios que encontraron un biomarcador que pueda utilizarse con este propósito, como por ejemplo el estudio retrospectivo de Fingerlin et al., en una pequeña cohorte, en el que los resultados poco concluyentes con la terapia antioxidante se vincularon a una variante del gen TOLLIP [15]. No podemos negar la importancia de la medicina de precisión como un objetivo en el tratamiento de la FPI, pero la esperanza de que biomarcadores, vinculados a las vías fisiopatológicas de la FPI puedan identificar subgrupos de pacientes que responden selectivamente a terapias dirigidas, sigue siendo hasta la actualidad solo un anhelo.
Por el otro lado, aparecen los “Agrupadores” de la FPI con otras enfermedades fibróticas: el fenotipo fibrótico progresivo. Este grupo de expertos cree que en lugar de desglosar el estudio de la FPI debemos intensificar nuestros esfuerzos en agrupar la FPI con otras EPID de causas conocidas o asociadas y con las idiopáticas, que poseen un comportamiento biológico, clínico y evolutivo similar [2,16]. El argumento de los “agrupadores” es que de esta forma generamos definiciones claras sobre un gran grupo de enfermedades fibrosantes y progresivas hasta ahora huérfanas, que permitirán desarrollar ensayos clínicos, conocer su comportamiento longitudinal y ofrecer un tratamiento basado en estudios serios.
Posiblemente el futuro esté en la medicina de precisión, pero creemos que el enfoque de aquellos que dividen la FPI de enfermedad pulmonar progresiva no FPI sigue siendo relativamente prematuro. Más allá de “cómo” llegan estos pacientes a la Fibrosis pulmonar, el destino parece ser uno solo, y esto es por ahora de lo que estamos seguros.
- Patogenia de las EPID fibrosantes con Fenotipo Progresivo
Desde un punto de vista fisiopatológico, la fibrosis resulta casi universalmente de la pérdida irreparable de integridad de la barrera epitelial o, en menor grado endotelial, observada en muchas de las EPID no FPI. Aunque la etiología de la injuria celular puede variar, ya sean mecanismos autoinmunes en enfermedades intersticiales asociadas a enfermedad del tejido conectivo (EPID-ETC), partículas de polvo orgánico inhalado en neumonitis por hipersensibilidad o una lesión desconocida en la FPI, es probable que el inicio de la respuesta fibrótica esté relacionado con la injuria persistente y mecanismos de reparación aberrantes de las células epiteliales y/o endoteliales. Del mismo modo, aunque los mecanismos de iniciación de la enfermedad puedan diferir de los de progresión, es muy probable que un espectro de mecanismos biológicos, incluida la activación del factor de crecimiento y la reprogramación epigenética de los fibroblastos determinen la probabilidad de progresión de la enfermedad en diversas EPID, a pesar de diferentes etiologías subyacentes y vías patológicas inicialmente distintas [2].
Los fibroblastos, las principales células efectoras en las EPID fibrosantes, se reclutan y se activan para convertirse en miofibroblastos, que secretan cantidades excesivas de matriz extracelular, lo que aumenta la rigidez del tejido y la pérdida de la función del tejido alveolar. Además, los macrófagos y los linfocitos se reclutan en el sitio de la lesión y liberan mediadores pro-fibróticos que promueven aún más la activación de fibroblastos. En un ciclo de feed-forward (prealimentación), el incremento de la rigidez del tejido pulmonar activa y estimula aún más los fibroblastos, en un proceso conocido como mecano-estimulación, para impulsar la fibrosis pulmonar progresiva y auto-sostenida [17]. (Fig. 2)
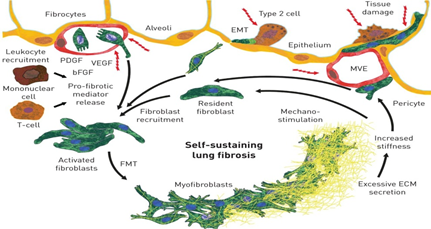
Figura 2: Mecanismos involucrados en la patogénesis y progresión de las enfermedades pulmonares intersticiales fibrosante. EMT: transición epitelial-mesenquimal; PDGF: factor de crecimiento derivado de plaquetas; VEGF: factor de crecimiento endotelial vascular; bFGF: factor de crecimiento básico de fibroblastos; MVE: epitelio microvascular; FMT: transición fibroblastos-miofibroblastos; ECM: matriz extracelular [17].
- ¿Cómo definimos progresión?
Ahora que hemos reconocido la presencia de este grupo de entidades fibrosantes progresivas, resulta fundamental definir que entendemos por progresión.
Antes que nada, hay que dejar en claro que “fibrosante progresivo” implica que el evento ha ocurrido a pesar de un tratamiento instaurado de acuerdo con los estándares actuales (corticoides y/o inmunosupresores). Muchas de estas enfermedades, tienen grados variables de inflamación y fibrosis en el parénquima pulmonar, y son pasibles de una eventual mejoría, al menos al inicio, con tratamiento antiinflamatorio. Si luego de indicar dicho tratamiento inmunosupresor evidenciamos empeoramiento, es ahí que debemos considerar la presencia de un fenotipo progresivo.
No existe consenso internacional aún para una definición unificada de fenotipo progresivo [18].
En la práctica clínica diaria, normalmente se utilizan una serie de medidas para monitorear la evolución de los pacientes con enfermedades fibrosantes pulmonares y la respuesta al tratamiento indicado. Estas medidas incluyen normalmente elementos clínicos (síntomas y tolerancia al ejercicio), de la función pulmonar (capacidad vital forzada y capacidad de difusión de monóxido de carbono), y radiológicos (empeoramiento de las opacidades fibróticas en la TCAR).
El deterioro de la calidad de vida cuantificado objetivamente mediante cuestionarios validados, las hospitalizaciones no programadas, los empeoramientos de la disnea en cortos periodos de tiempo y el inicio de oxigenoterapia ambulatoria, son también parámetros muy relevantes para el paciente y podrían ser usados para evaluar progresión y el efecto de las terapias instauradas.
En términos generales, la herramienta más usada para evaluar progresión de la enfermedad es la CVF y sus modificaciones a lo largo del tiempo, y puede ser acompañada en algunos de casos por cambios en la TCAR o en los síntomas, especialmente cuando los cambios en la CVF son marginales y pueda haber duda por la variabilidad del método de medición. Se debe tener presente también que la CVF puede hallarse pseudo-normalizada en pacientes con comorbilidad de enfisema y fibrosis pulmonar [19].
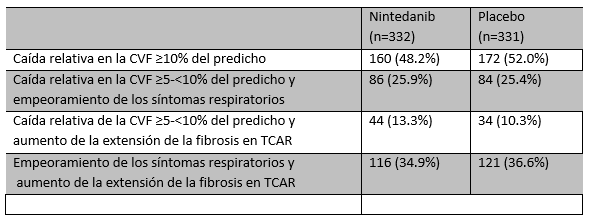
En ausencia de una definición normatizada internacionalmente, en general se acepta que los pacientes con una caída absoluta de ⩾10% en la FVC, o caída de ⩾15% in DLCO [3], o caídas marginales de entre el 5-10% de la FVC junto a empeoramiento de los síntomas o de las imágenes torácicas (TCAR), dentro de un período de 24 meses, han experimentado una progresión de su enfermedad fibrosante pulmonar [20]. Unos criterios similares fueron usados en el ensayo clínico INBULD, para evaluar progresión y elegibilidad para ingresa al estudio [21] (Ver Tabla 1).
Es necesario tener presente que al día de hoy es imposible predecir el curso de la enfermedad pulmonar fibrosante en un paciente individual, y esa capacidad vendrá en un futuro integrando biomarcadores de la genética y la biología molecular a la información que aportan la clínica, la función pulmonar y las imágenes.
| Tabla 1. INBUILD: Criterios de progresión en los 24 meses previos [22] |
La radiografía de tórax y por sobre todo la Tomografía Computada de Alta Resolución (TCAR), son componentes esenciales en el proceso diagnóstico y manejo de las enfermedades intersticiales pulmonares [22]. Ambas son consideradas técnicas no invasivas y pueden aportar información relevante, sobre todo dada la posibilidad de repetirse en el tiempo, obteniendo imágenes seriadas, que permiten una mejor valoración evolutiva del proceso fibrosante. La comparación con estudios previos puede ser crucial para la identificación de una enfermedad fibrosante progresiva, tanto en la radiografía convencional como en la TCAR.
La sensibilidad y especificidad de la radiografía de tórax es considerada inferior a la de la TCAR en el diagnóstico y pronóstico de los pacientes con enfermedades intersticiales. Una radiografía de tórax “normal”, no excluye la presencia de enfermedad intersticial en un contexto clínico adecuado [23]. Si bien la radiografía de tórax presenta baja especificidad y sensibilidad es ampliamente usada como método inicial de aproximación en pacientes con disnea con el fin de descartar otras patologías como, por ejemplo: infección, insuficiencia cardíaca o cáncer.
La TCAR es la modalidad por imágenes más sensible para la detección de anormalidades asociadas a enfermedades pulmonares intersticiales difusas. Hoy en día se considera que su rol en el diagnóstico de estas es central, ya que permite visualizar signos tempranos y muchas veces ayuda a definir patrones específicos, guiando la toma de decisiones dentro del algoritmo diagnóstico [24].
Además del rol diagnóstico, la TCAR se considera como herramienta pronóstica ya que la extensión de panalización y reticulación, como así también la severidad de las bronquiectasias por tracción, han sido descritos como predictores de mortalidad [25,26].
Un papel no menor es la monitorización del tratamiento, de las complicaciones y la detección de comorbilidades asociadas, como por ejemplo el cáncer de pulmón.
Asimismo, a través de las imágenes seriadas es posible identificar cambios en la extensión de los signos fibróticos clásicos (panalización, reticulación y bronquiectasias por tracción), permitiendo la identificación de pacientes con fenotipos fibrosantes progresivos que se correlacionan con peor sobrevida [27,28]. Sin embargo, es importante saber que es posible estar frente a un fenotipo fibrosante progresivo, demostrado por el deterioro clínico y de los estudios funcionales pulmonares con una aparente estabilidad en la tomografía computada.
En relación con esta última aseveración, creemos que la TCAR tiene sus limitaciones y que probablemente su combinación con otros biomarcadores sea una herramienta mucho más poderosa en el diagnóstico e identificación de enfermedades intersticiales fibrosantes potencialmente progresivas [29].
- Tratamiento de las EPID fibrosantes progresivas no-FPI
Debe tenerse en cuenta que el curso de la enfermedad para un paciente individual sigue siendo imposible de predecir. Hasta hace muy poco, no se contaba con ningún tipo de evidencia sobre el tratamiento de este tipo de pacientes con EPID fibrosantes progresivas; pero recientemente se han publicado los resultados de dos estudios: INBUILD [22] y uILD [30].
Estudio INBUILD
Es un estudio Fase III diseñado para identificar la eficacia del nintedanib en pacientes con EPID-FP. Se incluyeron pacientes mayores de 18 años con diagnóstico de EPID no-FPI que presentaran elementos radiológicos de enfermedad intersticial con una extensión mayor del 10% en la TCAR; CVF ≥45% del valor predictivo y DLCO entre 30% y 80%. Además debía objetivarse progresión mediante al menos uno de los criterios descriptos en Tabla 1, en los 24 meses previos a la aleatorización. Como criterio de valoración o desenlace primario se tuvo en cuenta la tasa anual de descenso de la CVF (ml/año) evaluada a lo largo de 52 semanas. Las características basales de los pacientes eran similares en las dos ramas, nintedanib y placebo y el 62% de los pacientes presentaban patente de NIU en la TCAR. Con respecto a los diagnósticos, el 25.6% de los pacientes presentaba neumonitis por hipersensibilidad crónica, el 25.6% enfermedad del tejido conectivo (el 13.34% artritis reumatoidea) y el 18.8% neumonía intersticial no especifica idiopática. Resultados. Globalmente el tratamiento con nintedanib se asoció con una reducción relativa de la tasa anual de declinación de la CVF del 57% versus placebo. El perfil de eventos adversos del nintedanib en el estudio INBUILD fue similar al de los estudios INPULSIS y SENSCIS. Los eventos adversos más frecuentes fueron los gastrointestinales. No surgieron alertas nuevas de seguridad con el uso de nintedanib.
Estudio uILD
Es un estudio Fase II, multicéntrico, diseñado para evaluar seguridad y eficacia del tratamiento con pirfenidona versus placebo en pacientes con EPID-FP. Se incluyeron pacientes entre 18 y 85 años, con enfermedad intersticial pulmonar inclasificable con CVF ≥45%, DLCO ≥30%, compromiso fibrosante en la TCAR mayor del 10% y progresión documentada en los 6 meses previos. Se aleatorizaron 253 pacientes (127 recibieron pirfenidona a dosis de 2403 mg diarios y 126 placebo). El desenlace primario fue el cambio medio en la capacidad vital forzada en mililitros a las 24 semanas medido diariamente mediante un espirómetro domiciliario. Como desenlaces secundarios se plantearon los cambios en la CVF medida con un espirómetro en el consultorio, proporción de pacientes con un descenso absoluto o relativo mayor de 5% y de 10% en la declinación de la CVF, cambios en la DLCO medida en porcentaje del predictivo y cambios en la distancia recorrida en la prueba de la marcha de los 6 minutos en cada visita médica; cambios en los cuestionarios de calidad de vida USCSD-SOBQ, SGQR; proporción de pacientes no hospitalizados, exacerbaciones agudas y supervivencia libre de progresión. Las características basales de la población eran similares entre las dos ramas. La mayoría de los pacientes presentaban diagnóstico de enfermedad intersticial inclasificable sin elementos sugestivos de otra forma de enfermedad intersticial. Resultados. El análisis del desenlace primario, como estaba pre-especificado en el plan de análisis estadístico, fue imposible de realizarse debido a cuestiones de confiabilidad técnica, referentes a las espirometrías domiciliarias grabadas en los espirómetros. A la semana 24 la declinación media de la CVF fue menor en los pacientes tratados con pirfenidona que en el grupo placebo (diferencia entre grupos de 95,3 ml p=0·002). Del total de pacientes con DLCO y prueba de la marcha disponibles a la semana 24, el cambio medio en el porcentaje del valor predictivo de la DLCO desde basal fue de –0,7% para la rama pirfenidona (n=97) y –2,5% para la rama placebo (n=110). La media del cambio en la distancia recorrida en la PM6M desde el valor de base fue de –2,0 m para el grupo pirfenidona (n=99) y –26,7 m para el grupo placebo (n=108). Con respecto a los cuestionarios aplicados no se encontraron diferencias significativas entre las dos ramas, así como en presentación de exacerbaciones agudas, hospitalizaciones y tiempo hasta la muerte por causas respiratorias. Tampoco se encontraron diferencias en la supervivencia libre de progresión de la enfermedad. Los eventos adversos más frecuentes relacionados con el tratamiento fueron gastrointestinales, fatiga y rash. En conclusión, aunque el modelo estadístico planificado para este estudio no pudo ser aplicado para el análisis de los datos del desenlace primario, el análisis de los desenlaces secundarios clave sugiere que los pacientes con enfermedad pulmonar intersticial inclasificable fibrosante progresiva podrían beneficiarse del tratamiento con pirfenidona. En este estudio el tratamiento con pirfenidona mostró un perfil de seguridad y tolerancia aceptable.
- Conclusiones
Un grupo de pacientes con EPID fibrosantes no FPI desarrollan un fenotipo progresivo (EPID-FP) caracterizado por la declinación en la función pulmonar, mayor compromiso en la TCAR, empeoramiento de la calidad de vida y finalmente muerte precoz. Si bien no existe consenso en cómo se debe definir la progresión de la enfermedad en estos pacientes, la evaluación de la función pulmonar, síntomas, imágenes y eventos como las exacerbaciones agudas y hospitalizaciones son de gran valor en la práctica clínica. Los estudios con nintedanib y pirfenidona en las EPID-FP demostraron que su efecto enlentecedor de la fibrosis podría extenderse a otras enfermedades intersticiales fibrosantes no FPI. Evidentemente se abren nuevas perspectivas. Quizás seamos contemporáneos a un cambio de paradigma en el diagnóstico, tratamiento y seguimiento de las EPID no-FPI [9].
- Bibliografía
- Flaherty KR, Brown KK, Wells AU et al. Design of the PF-ILD trial: a doubleblind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res 2017; 4(1): e000212.
- Wells AU et al. What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J 2018; 51(5):1800692.
- Du Bois RM, Weycker D, Albera C et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med 2011; 184:1382–1389.
- Cottin V et al. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev 2019; 28(151):180100.
- Tzouvelekis, A., Bouros, D. Endotyping of progressive fibrotic interstitial lung diseases: It is the final destination that matters and not the journey. EBioMedicine 2020; 51.
- Hoffmann-Vold AM et al. Tracking impact of interstitial lung disease in systemic sclerosis in a com- plete nationwide cohort. Am J Respir Crit Care Med 2019 Epub ahead of print.
- Hoffmann-Vold, A. M.etal. Endotype–phenotyping may predict a treatment response in progressive fibrosing interstitial lung disease. EBioMedicine 2019; 50, 379-386.
- Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J RespirCrit Care Med 2018;198(5):44–68
- Collins B. F., Raghu G. Antifibrotic therapy for fibrotic lung disease beyond idiopathic pulmonary fibrosis. European Respiratory Review 2019; 28(153), 190022.
- Spagnolo, P., &Cottin, V. Genetics of idiopathic pulmonary fibrosis: from mechanistic pathways to personalised medicine. Journal of medical genetics 2017; 54(2), 93-99.
- Mathai, S. K., Schwartz D. A. 2015. Taking the “I” out of IPF. Eur Respir J 2015;45:1539-1541
- Wolters, P. J. et al Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? The Lancet. Respiratory medicine 2018; 6(2), 154–160.
- Noth, I. et al Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. The lancet Respiratory medicine 2013; 1(4), 309-317.
- Peljto, A. L., Zhang, Y., Fingerlin, T. E., Ma, S. F., Garcia, J. G., Richards, T. J., …& Gibson, K. F. (2013). Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. Jama, 309(21), 2232-2239
- Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 2013; 45: 613–620
- Walsh SL, Richeldi L. Subclinical interstitial lung abnormalities: Lumping and splitting revisited. Am J Respir Crit Care Med.2019;200(2):121-123
- Wollin L. et al. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. European Respiratory Journal 2019; 54(3), 1900161.
- Cottin V. Treatment of progressive fibrosing interstitial lung diseases: a milestone in the management of interstitial lung diseases. European Respiratory Review 2019;28:190109
- Cottin V et al. Effect of emphysema extent on serial lung function in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(9):1162–71.
- Flaherty KR, Brown KK, Wells AU et al. Design of the PF-ILD trial: A double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res. 2017;4(1).
- Flaherty KR, Wells AU, Cottin V et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–27.
- Travis WD, King TE, Bateman ED et al. American thoracic society/European respiratory society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. In: American Journal of Respiratory and Critical Care Medicine. American Lung Association 2002; 277–304.
- Ryu JH, Daniels CE, Hartman TE, Yi ES. Diagnosis of interstitial lung diseases. Vol. 82, Mayo Clinic Proceedings. Elsevier Ltd 2007; 976–86.
- Raghu G et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J RespirCrit Care Med. 2011; 183(6):788–824.
- Lynch DA et al. High-resolution computed tomography in idiopathic pulmonary fibrosis: Diagnosis and prognosis. Am J RespirCrit Care Med. 2005; 172(4):488–93.
- Walsh SLF et al. Connective tissue disease related fibrotic lung disease: High resolution computed tomographic and pulmonary function indices as prognostic determinants. Thorax. 2014; 69(3):216–22.
- Walsh SLF et al. Role of imaging in progressive-fibrosing interstitial lung diseases. EurRespir Rev. 2018; 27(150).
- Oda K, Ishimoto H, Yatera K, Naito K, Ogoshi T, Yamasaki K, et al. High-resolution CT scoring system-based grading scale predicts the clinical outcomes in patients with idiopathic pulmonary fibrosis. Respir Res. 2014; 15(1):10.
- Ley B et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012; 156(10):684–95.
- Maher T, Corte T, Fischer A, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med 2019 Published Online September 29, 2019 https://doi.org/10.1016/ S2213-2600(19)30341-8.