APORTE. Sección Fibrosis Quística. Coordinadora: Dra. Patricia Andreozzi.
AUTOR: Dra Patricia Andreozzi. Neumonóloga Infantil.
COLABORADORAS:
Dra. Laura Osken. Neumonóloga Adultos.
Dra. Silvina Smith. Neumonóloga Infantil.
Dra. Gladys Kahl.Neumonóloga Adultos
Dra. Eugenia Alais. Neumonóloga Adultos
La Fibrosis Quística (FQ) es una enfermedad genética con patrón de herencia autosómica recesiva. Está producida por mutaciones en el gen que codifica la proteína reguladora de la conductancia transmembrana CFTR; que funciona como un canal regulando del transporte de cloro y que se expresa en la membrana apical de células epiteliales con una afectación multisistemica.
El Diagnóstico precoz de la enfermedad aumenta la expectativa de vida de los pacientes.
A -Herramientas diagnósticas que confirman la enfermedad:
La evaluación de la disfunción del CFTR es fundamental para definir el diagnóstico y se objetiva mediante: 1) La prueba o Test del sudor con determinación anormal de cloruro.2) Identificación de mutaciones causantes de FQ (definidas en la base de datos CFTR2) 3) ó Análisis funcional CFTR, midiendo la Diferencia de potencial nasal (DPN) o medición de la corriente intestinal (MCI) cuyos valores son anormales.
1-Test del Sudor Gold Estándar para el Diagnóstico: confirma la enfermedad cuando un paciente presenta síntomas compatibles, pesquisa neonatal positiva o historia familiar de FQ, demuestra una concentración de cloruro igual o superior a 60 mmol/L (FQ confirmada); es probable la FQ, si los valores se encuentran en límites intermedios de cloruros (Cloruros 30 a 59 mmol/L) e improbable de tener FQ (Cloruros debajo de 30 mmol /L).
Existen algunas mutaciones del gen CFTR, como c.3717 + 12191C> T (antes denominada 3849 + 10 kb C-> T), están asociadas con valores bajos de cloruros en el sudor.
El Test del sudor determina la concentración de cloruros en el sudor. La única técnica universalmente aceptada es la descrita por Gibson y Cooke.; esta técnica de iontoforesis con estimulación con pilocarpina, requiere de una metodología rigurosa para su realización, para evitar los resultados falsos positivos y falsos negativos.
Interpretación de los resultados
Normales Menor de 30 mmol/L
Intermedio Intermedio 30 a 59 mmol/L
Patológicos Mayor de 60 mmol/L
Cantidad de sudor mayor a 100 mgr.
Peso del lactante mayor de 2 kg .
A partir de los 15 días de vida
Test del Sudor con valores intermedios se deben repetir en laboratorios con experiencia y derivar a Centros de Atención especializados para seguimiento de pacientes con F.Q.
2-Biologia Molecular. El hallazgo de dos mutaciones relacionadas con la FQ, en ambas copias del gen CFTR, confirmaría el diagnóstico de la enfermedad. Se han descrito más de 2000 mutaciones, aunque no en todos los casos se ha establecido bien su relación con la producción de enfermedad. En caso de duda se puede consultar la página web del Consorcio Internacional para el análisis genético de la FQ http://www. genet.scikkids.on.ca/cftr2/.
3- Diferencia de Potencial nasal y Medición de la corriente intestinal: El transporte de iones en el líquido periepitlial genera una diferencia de potencial transepitelial que resulta diferente en los pacientes con FQ. Las pruebas son confiables, pero poco accesibles por lo que son de uso limitado.
B- Pesquisa neonatal
Con el objeto de prevenir la detección tardía de la enfermedad, fueron creados los programas de pesquisa o screening neonatal que al ser aplicados rápidamente se puede sospechar la enfermedad y luego confirmarla antes que los síntomas aparezcan.
La Tripsina Inmunoreactiva (TIR) forma parte de la pesquisa neonatal y es una herramienta de sospecha de F.Q; cuyos valores se encuentran aumentados en las primeras 48 a 72hs de nacimiento en pacientes con F.Q
Existen diferentes protocolos de Pesquisa Neonatal en Fibrosis Quística:
1-TIR/TIR. El segundo TIR se debe realizar entre 2 y 3 semana de vida. Después del mes la prueba no tiene validez. En caso de ser elevado, el diagnóstico de F.Q se confirmará con Test del Sudor y/o Biología Molecular. Sensibilidad menor de 83% y valor predictivo positivo 10 a 20%
2-TIR /PAP.( Proteina asociada a la Pancreatitis) parece ser más eficiente que el TIR/TIR, porque reduce el tiempo para la confirmación de la enfermedad en caso de pesquisa positiva y no requiere recitación del RN. Sensibilidad de 80 a 100% y valor predictivo positivo de 4 a 8%
3-TIR /ADN: Alto Valor predictivo, pero tienen la desventaja de detectar portadores sanos y mutaciones sin valor clínico.
Ningún protocolo de pesquisa es ideal y puede tener falsos negativos y positivos
En nuestro país, la implementación del Programa Nacional de Pesquisa Neonatal nos enfrenta a nuevos desafíos diagnósticos de la enfermedad, y considerando que en algunas provincias a nivel público y privado no se han implementado adecuadamente las estrategias de doble tripsina inmunoreactiva TIR/TIR, ó TIR/PAP y TIR/ADN se dificulta aún más la detección y diagnóstico temprano de esta enfermedad. En el Registro Argentino de Fibrosis Quística del año 2018 se publicó 1159 casos. En el grupo de niños de menores de 1 año, el diagnóstico fue a partir de la sospecha por pesquisa neonatal positiva en un 67 %.
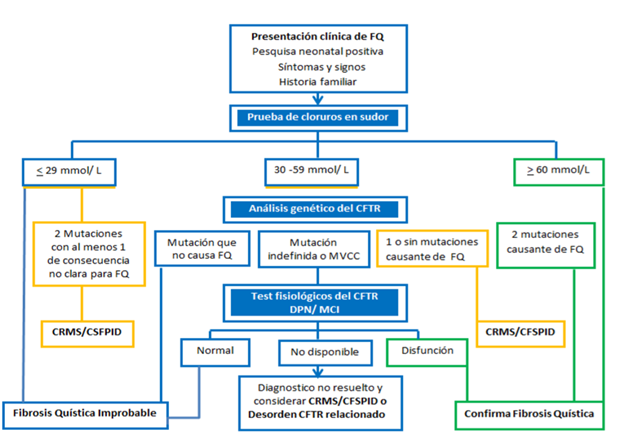
Algoritmo Diagnóstico
Cuando el paciente tiene pesquisa positiva y/o síntomas y /o signos clínicos (Una o más de las siguientes características fenotípicas: síntomas respiratorios; síndrome malabsortivo; deshidratación; edema; anemia; ileo meconial; azoospermia; etc y/o historia familiar de F.Q
1 – Se debe Confirmar el diagnóstico de F.Q con: 1 test del sudor patológico y/o el hallazgo de 2 mutaciones genéticas causante de enfermedad.
2 -Si el test de sudor es normal y no existen alteraciones genéticas es improbable el diagnóstico de Fibrosis Quística.
3- Un gran problema se plantea si el test del sudor presenta valores intermedios, con estudios genéticos, y potenciales nasales no definitorios. Denominamos a ésta situación: Diagnóstico de Fibrosis Quística no resuelta. También la situación de Solo Pesquisa positiva: diagnóstico Inconcluso: (CFSPID) y Trastornos relacionados con CFTR en los casos de: Azoospermia por ausencia de conductos deferentes /Pancreatitis recurrente / bronquiectasias sin afectación pancreatica). Estas últimas identidades clínicas también requieren seguimiento en un Centro Especializado para Fibrosis Quística.
Tener en cuenta que la clínica es soberana y el paciente a pesar de no tener los estudios definitorios es FQ !!!!.
La Fibrosis Quística constituye un verdadero desafío diagnóstico desde el RN a la adultez; la enfermedad evoluciona en sus diferentes formas clínicas de leves a severas.
En la actualidad existe una verdadera ventana de posibilidades diagnósticas y terapéuticas y no faltaría mucho para la cura, que se encuentra ya en etapa de investigación.
Philip M. Farrell, MD, PhD. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. The Journal of Pediatrics. 2017 JOURNAL OF PEDIATRICS